[Edited to simplify the question]
I am trying to associate the eigenvalues $E$ of a matrix $H$ to the original rows of the matrix.
Moreover, it would be trivial to sort the eigenvalues in ascending order, however this throws away some of the information. For my purpose the matrix is a Hamiltonian where the different rows have physical meaning. I always want the eigenvalue that corresponds to a given row.
I have tried to sort the eigenvalues according to the dominant contribution. To do this I get the magnitudes of the eigenvectors (they are complex) for each row and pick the largest. I use this index to sort the eigenvalues.
# Get eigenvalues and vector of the Hamiltonian
E, V = eig(H)
# Now sort the eigenvalues by dominant contribution
sorted_order = []
for i in range(len(E)):
row = V[i,:]
row = np.absolute(row) # length of complex number <-- correct?
largest_index = np.argmax(row) # the index of the largest value
sorted_order.append(largest_index)
E_sorted = E[sorted_order]
The sorting approach works fairly well when the eigenvector matrix is dominated by the diagonal. However, if the matrix is more homogeneous the this sorting method fails.
This leads me to believe that my method of sorting is probably incorrect. Or I am running in to accuracy issues with the eig function.
Does anyone have suggestion on an alternative way to sort by the dominant contribution?
Background
I am doing a $\mathbf{k}\cdot\mathbf{p}$ simulation for semiconductor band structure. The eigenvalues for the $\mathbf{k}\cdot\mathbf{p}$ Hamiltonian (the 8x8 matrix below) corresponds to the different semiconductor bands and the eigenvectors are the wavefunctions.

(NB I have removed two rows and columns because I am not interested in their effect for by purpose.)
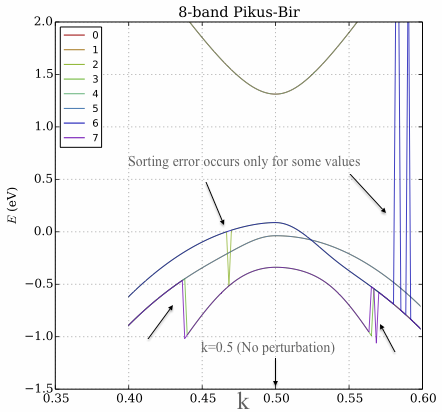
Figure: In the plot the y-axis is the eigenvalues, the x-axis corresponds to a k-space vector which changes values in the Hamiltonian. A definition: at $k=0.5$ the system is unperturbed and the eigenvectors are dominated by the diagonal terms. This also illustrates why I cannot do a simple energy sort: because at some points the bands cross. The bands have different physical meaning which I want to preserve.
Comment. The sorting approach (the Python code above) works fairly well when $k=0.5$ because the contribution is dominated by the diagonal of the eigenvectors. However, moving away from the centre of k-space ($k>0.5$ and $k<0.5$) the eigenvector matrix becomes more homogeneous and this method of sorting fails. I am surprised that it is failing intermittently.